|
|
|
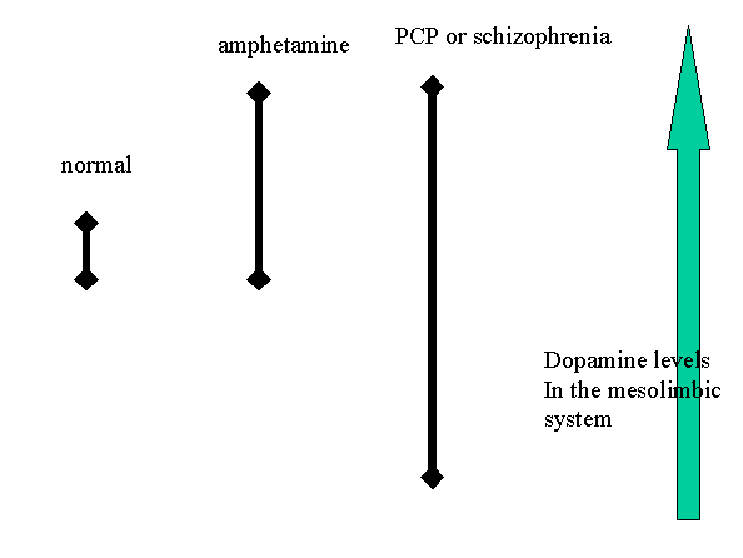
SchizophreniaSchizophrenia has acute symptoms like hallucinations, speech incoherence and delusions, and chronic symptoms like apathy, alogia (speech impairment), affective flattening, attentional impairment, and antisociability. The chronic symptoms are unresponsive to drug therapy, implying they’re due to structural changes like ventricular enlargement in the brain. The acute symptoms are responsive to drug therapy, implying they are due to dopamine dysfunction. Evidence for this includes the fact that dopamine agonists like amphetamine can induce schizophrenic-like symptoms, e.g. amphetamine psychosis is like paranoid schizophrenia. Amphetamine causes stereotypy in rats, repetition of complex non-adaptive behaviours, like throwing food around. This is a disorder of perception thought to parallel schizophrenic disorders of thought. In monkeys it can cause visual hallucinations, e.g. catching invisible flies, thought to parallel the auditory hallucinations in schizophrenia. Amphetamine makes people showing chronic symptoms of schizophrenia better, perhaps by kick-starting activity, motivation, positive affect, but makes people with acute symptoms worse. Dopamine is a precursor of norepinephrine, but it’s not norepinephrine that’s important. Evidence this includes the fact that fusaric acid blocks the conversion of dopamine to norepinephrine, and administering this to patients makes them worse. Giving patients levodopa, a precursor to dopamine, and fusaric acid worsens symptoms more. Typical antipsychotics, like the phenolthiazine chlorpromazine, block the production of dopamine secondary messenger systems such as cAMP and adenylate cyclase. Chlorpromazine works by blocking the dopamine receptors. Although this tells the neuron “there’s no dopamine out there” thereby increasing dopamine production, this has no effect because the receptors are blocked. Antipsychotic drugs such as chlorpromazine are thought to exert their therapeutic effects by blocking dopamine receptors. Chlorpromazine is injected first and occupies postsynaptic receptor sites. Then amphetamine is injected. This mimics the arrival of an action potential at the nerve ending and causes the release a large number of DA molecules from the presynaptic neuron. But this released DA cannot activate the postsynaptic receptors because they are occupied by chlorpromazine molecules. Consequently, the postsynaptic neuron does not generate an action potential. The released DA is broken down by enzymes in the synaptic cleft, or is reabsorbed into the presynaptic neuron.
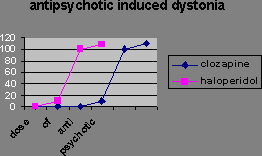
There are 2 types of dopamine receptor. Evidence for this is that some antipsychotics, e.g. the phenolthiazine chlorpromazine increase production of adenylate cyclase, while others, e.g. the butyrophenones spiperone and haloperidol, don’t. Activation of D1 receptors increases adenylate production; activation of D2 receptors inhibits it. The dopaminergic dysfunction is at the D2 receptor. Evidence for this includes the fact that the inhibition of haloperidol binding is proportional to drug therapeutic effect. Spiperone binds more to the nucleus accumbens and striatum of schizophrenic patients than controls. The thioxanthine flupenthixol binds more to the D2 receptors of schizophrenic patients than the D1. The number of D2 receptors upregulates in schizophrenics. Note these patients had all been on medication. Side effects of the typical antipsychotics are extrapyramidal effects ("extrapyramidal system" refers to the basal ganglia and brain stem nuclei with which they are connected), due to the fact the drug effect is not localised so you get Parkinson’s-like symptoms because dopamine effect has also been blocked in the nigrostriatal dopaminergic system. Side effects include: akathesia (motor restlessness), dystonia (prolonged muscle twitching), and tardive dyskinesia (spasms of facial muscles). Though the Parkinson’s-like symptoms disappear when you stop using the drug, the tardive dyskinesia may be more permanent. It’s also seen on animals using haloperidol as vacuous mouth movements. It’s due to dopamine receptor hypersensitivity because the antipsychotics block the receptors. Atypical antipsychotics, e.g. clozapine: (1) reduce or leave unchanged prolactin levels (as dopamine normally inhibits serum prolactin production, this is a measure of D2 receptor blockade), (2) have few or no extrapyramidal symptoms, (3) work on chronic or cognitive symptoms. Atypicals do induce extrapyramidal side effects but at much larger doses than that required for therapeutic effect. The therapeutic window is larger:
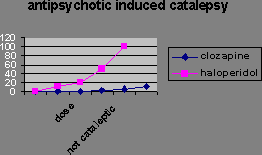
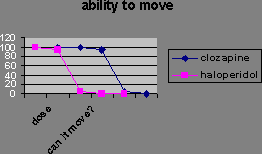
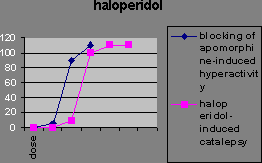
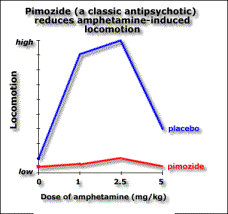
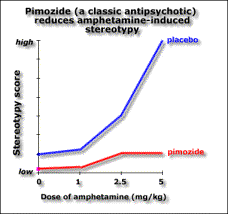
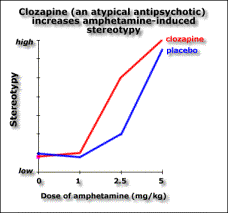
You can distinguish them in animals because they reduce amphatamine-induced “schizophrenia” without stopping the animals moving around. Catalepsy is a difficulty initiating movement, a typical effect of administering a dopamine antagonist.
Extrapyramidal side-effect separation is the dose range where the “psychotic” symptoms are suppressed, but most of the animals do not show catalepsy.
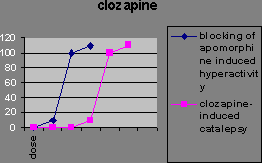
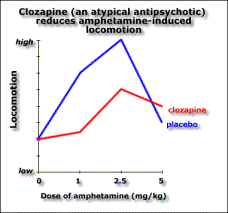
The atypical antipsychotics, such as clozapine, don’t block apomorphine-induced stereotypy. The ability to block stereotypy may indicate the drug has side effects.
This implies that if the drug blocks stereotypy, it’ll cause side effects. Evidence for this is the fact that Parkinson’s and catalepsy are both related to reduced postsynaptic dopamine activation in the striatum. Typical antipsychotics have a blocking effect here. Dopamine induced stereotypy and hyperactivity is due to increased activity here (and the nucleus accumbens). So we don’t want the drug to reduce dopaminergic action in the striatum, hence stereotypy should remain unchanged in order not to get catalepsy. The therapeutic effect of the drug may be due to increased activity in the nucleus accumbens, thus affecting locomotion. Evidence for this is that (from in vivo microdialysis) clozapine and haloperidol both cause an increase in dopamine in the nucleus accumbens, a compensatory response measuring the blocking effect, though clozapine causes it selectively there while haloperidol also causes it in the striatum. Clozapine may also increase dopamine levels in the prefrontal cortex according to a contradictory study. If we lesion dopaminergic terminals in the medial prefrontal cortex, we get pre- and post- dopaminergic synapse receptor overactivity in the basal ganglia (striatum) and limbic areas (nucleus accumbens). So the prefrontal cortex normally inhibits striatal and limbic areas. So if the prefrontal cortex were impaired, say through ventricular enlargement (causing the negative symptoms), we’d get limbic and striatal hyperactivity causing the positive symptoms. Also, complete blocking is not as effective as partial blocking else you get upregulation of dopamine receptors. Upregulation is associated with the long-term effect of tardive dyskinesia, and lower therapeutic effect. Evidence for this is the weak binding of clozapine for D2 receptors compared to haloperidol. Clozapine reduces dopaminergic effect at D2 receptors. Clozapine upregulates D1 occupancy, but D1 occupiers don’t seem to have any therapeutic effect. Another theory has been that the lack of side effects might be due to atypical antipsychotics’ greater affinity to serotonin receptors. But serotonin antagonists don’t reduce side effects in general. Apart from clozapine whose therapeutic window seems to be related to D2 affinity, the size of the therapeutic window in other atypicals does seem to be related to their 5HT2 affinity. The final word of note is the importance of glutamate to the dopaminergic system. Dopamine is released into synapses either in a spike-dependent way in reaction to stimuli, or in a tonic spike-independent way due to glutamate release by nerves from the prefrontal cortex into the synapse. If the prefrontal cortex is impaired, then there is less tonic release of dopamine in the striatal and limbic areas, and phasic release upregulates, leading to larger phasic peaks. Evidence for this theory includes the fact that glutamate-agonist induced “psychosis” hyperactivity and stereotypy is responsive to all antipsychotics (not just typicals) so atypicals must act at glutamate receptors, clozapine increases glutamate in the nucleus accumbens while haloperidol doesn’t, and clozapine upregulates PCP binding (a compensatory mechanism). So negative schizophrenic symptoms are due to tonic innervation depression, while acute symptoms are due to phasic dopamine overactivity, and it’s here lower down that the antipsychotic drugs have effect.
|